# 一番簡単?な方法は`stats::heatmap()`を使う。
# パッケージをインストールする必要もない。
stats::heatmap(as.matrix(mtcars))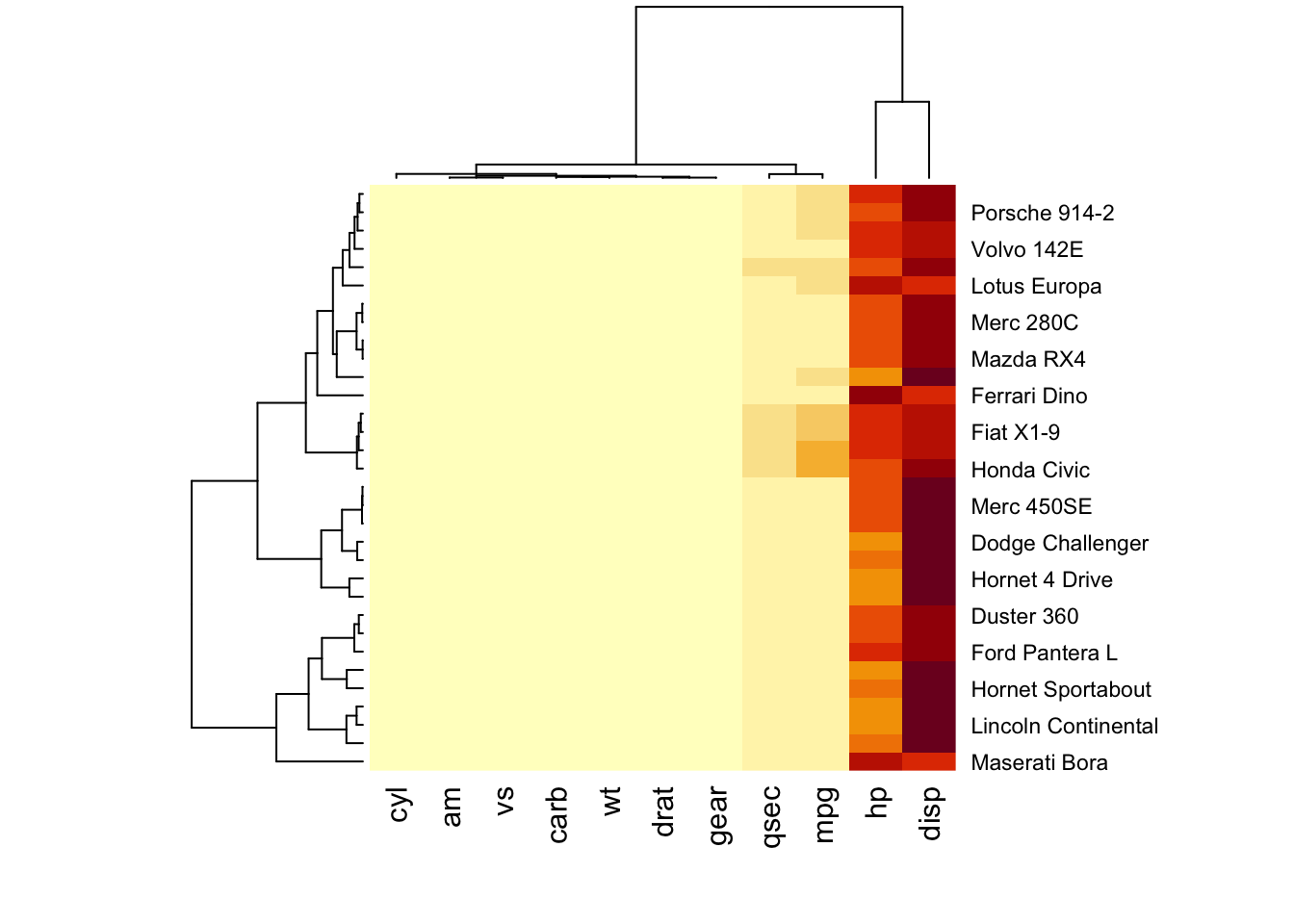
Rでヒートマップを作成するには、専用の関数やパッケージを使えばできる。
# 一番簡単?な方法は`stats::heatmap()`を使う。
# パッケージをインストールする必要もない。
stats::heatmap(as.matrix(mtcars))
他のヒートマップ作成用のパッケージもある。
pheatmapComplexHeatmapこれらの関数を使用する代わりに、 ggplot2の扱いに慣れていればヒートマップをggplot2でも描くことができる。 ggplot2でヒートマップを描く利点は、 他のパッケージや関数の使用方法を新たに勉強する必要がないこと、 そして自分でプロットを細かくコントロールすることができることにある。 ここでは、ggplot2を使ってヒートマップを描く方法について、 いくつかのテクニックと共に紹介する。
library(magrittr)
library(ggplot2)まずはヒートマップを作成したいデータをdata.frameにする必要がある。 多くの場合、x軸あるいはy軸に遺伝子やサンプル、処理条件などを配置し、 各セルの色を何らかの値(遺伝子発現量など)にするだろう。
ここでは横軸をサンプル、縦軸を遺伝子、 セルの色を遺伝子の各サンプルにおける遺伝子の発現量としたヒートマップを描くことにする。
デモシナリオとして、何らかの処理区と非処理区のサンプルについて、 RNA-seqを行ない、得られたリードカウントデータを元にヒートマップを描くことを考える。 簡易的なシミュレーションで、擬似リードカウントデータを生成して、 プロット作成に使う。 何度かパターンを変えてヒートマップを作成したいので、簡単にデータが生成できるように、 データ生成用の関数を作っておき、それを利用する。
# 遺伝子発現量のdata.frameの元を準備
make_tbl_exp_table <- function(n_gene = 1000, n_rep = 3) {
if(n_gene < 1) stop("n_gene should be more than 1.")
DIGIT <- floor(log10(n_gene)) + 1L
if(n_rep < 1) stop("n_rep should be more than 1.")
tbl_plot <-
tidyr::crossing(
gene = sprintf(paste0("gene%0", DIGIT, "d"), seq_len(n_gene)),
condition = c("control", "treat"),
rep = seq_len(n_rep)
) %>%
dplyr::mutate(exp = NA) %>%
tidyr::pivot_wider(names_from = c(condition, rep), values_from = exp)
tbl_plot
}
make_tbl_exp_table()# A tibble: 1,000 × 7
gene control_1 control_2 control_3 treat_1 treat_2 treat_3
<chr> <lgl> <lgl> <lgl> <lgl> <lgl> <lgl>
1 gene0001 NA NA NA NA NA NA
2 gene0002 NA NA NA NA NA NA
3 gene0003 NA NA NA NA NA NA
4 gene0004 NA NA NA NA NA NA
5 gene0005 NA NA NA NA NA NA
6 gene0006 NA NA NA NA NA NA
7 gene0007 NA NA NA NA NA NA
8 gene0008 NA NA NA NA NA NA
9 gene0009 NA NA NA NA NA NA
10 gene0010 NA NA NA NA NA NA
# ℹ 990 more rows# 基本の発現量を設定
mutate_base_exp <- function(tbl, seed = 777) {
set.seed(seed)
tbl %>%
dplyr::mutate(base = rnbinom(n = nrow(tbl), mu = 100, size = 1) + 1L,
.before = 2)
}
# 処理による発現変動(log2FC)を設定
mutate_log2fc <- function(tbl, seed = 777) {
set.seed(seed)
tbl %>%
dplyr::mutate(log2fc = rnorm(n = nrow(tbl), sd = 2), .after = base)
}
# 基本の発現量と発現変動量をもとに、各遺伝子の各サンプルにおける発現量を計算
assign_expression <- function(tbl, seed = 777) {
set.seed(seed)
tbl %>%
dplyr::mutate(dplyr::across(dplyr::starts_with("control"), \(x) {
purrr::map2_int(x, base, ~ rnbinom(n = 1, mu = .y, size = 1/.2))
})) %>%
dplyr::mutate(dplyr::across(dplyr::starts_with("treat"), \(x) {
purrr::map2_int(x, (2^log2fc)*base, ~ rnbinom(n = 1, mu = .y, size = 1/.2))
}))
}
# 関数を元に発現量のdata.frameを作成
tbl_exp <-
make_tbl_exp_table(n_gene = 200, n_rep = 5) %>%
mutate_base_exp() %>%
mutate_log2fc() %>%
assign_expression()
tbl_exp# A tibble: 200 × 13
gene base log2fc control_1 control_2 control_3 control_4 control_5 treat_1
<chr> <dbl> <dbl> <int> <int> <int> <int> <int> <int>
1 gene0… 87 3.78 117 43 48 110 146 1152
2 gene0… 212 -3.41 223 270 121 159 215 15
3 gene0… 157 0.617 196 274 106 195 356 553
4 gene0… 33 -0.259 16 25 25 50 23 36
5 gene0… 47 2.19 117 44 73 70 29 187
6 gene0… 73 0.788 85 46 33 126 113 122
7 gene0… 31 2.61 56 24 63 29 29 209
8 gene0… 210 2.33 133 186 194 251 160 2040
9 gene0… 34 -0.639 65 21 46 31 38 37
10 gene0… 83 2.17 28 98 102 84 100 273
# ℹ 190 more rows
# ℹ 4 more variables: treat_2 <int>, treat_3 <int>, treat_4 <int>,
# treat_5 <int>ここまで準備できたら、最低限の設定をしたヒートマップを作成していく。 基本的なコンセプトは非常に単純、
ggplot2で扱いやすいようにdata.frameを変形、変数(列)を追加geom_tile)を選択して、横軸にサンプル、縦軸に遺伝子、色に発現量を指定する。たったこれだけである。 ここでは、tidyr::pivot_longer()でdata.frameを変形して、 ggplot()でx軸にサンプルの列、y軸に遺伝子IDの列を指定し、 geom_tile()でセルの色に発現量の列を指定していく。
gp <-
tbl_exp %>%
tidyr::pivot_longer(
cols = !c(gene, base, log2fc),
names_to = "sample",
values_to = "readcount"
) %>%
ggplot(aes(x = sample, y = gene)) +
geom_tile(aes(fill = readcount + 1))
gp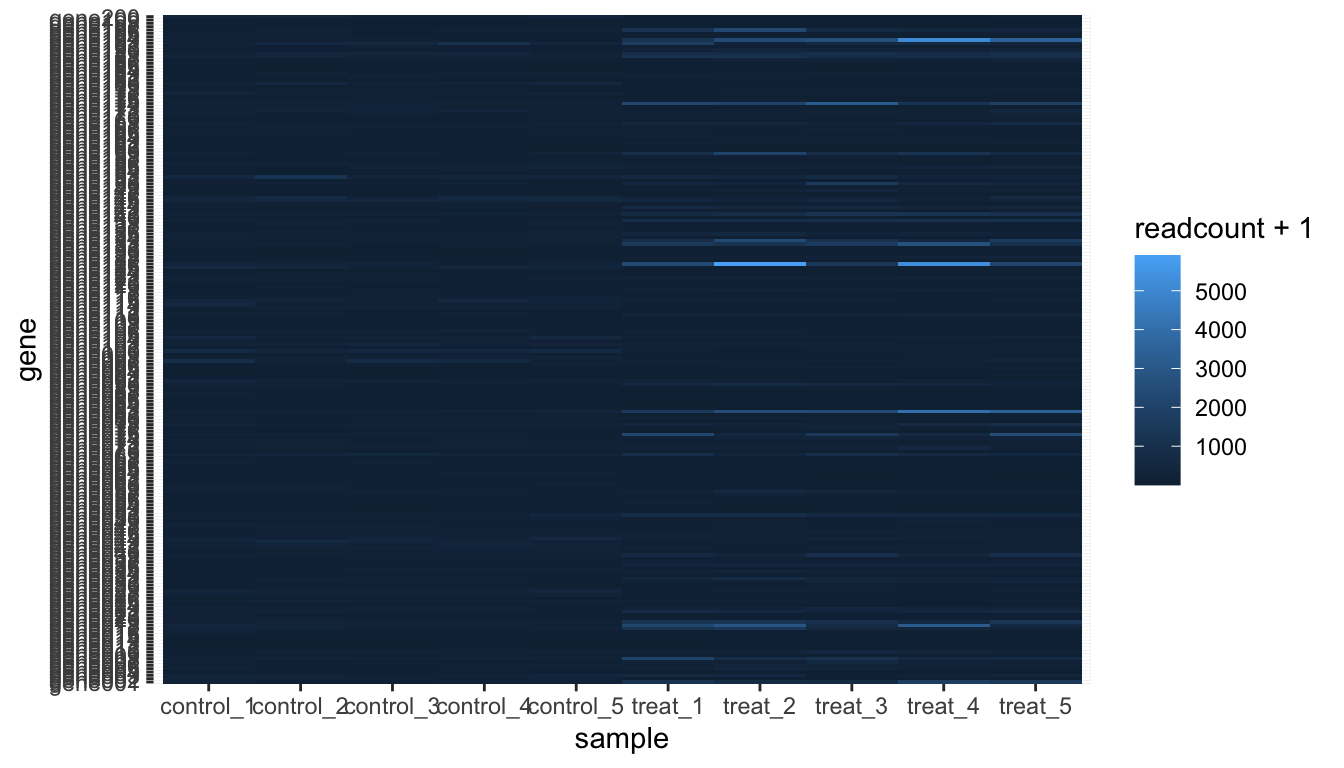
あとは、必要に応じてヒートマップの体裁を整えていく。
## 横軸のタイトルを消去
(gp <- gp + labs(x = ""))
## 発現量がリードカウントなので、log変換して色分け
(gp <- gp + scale_fill_continuous(trans = "log10"))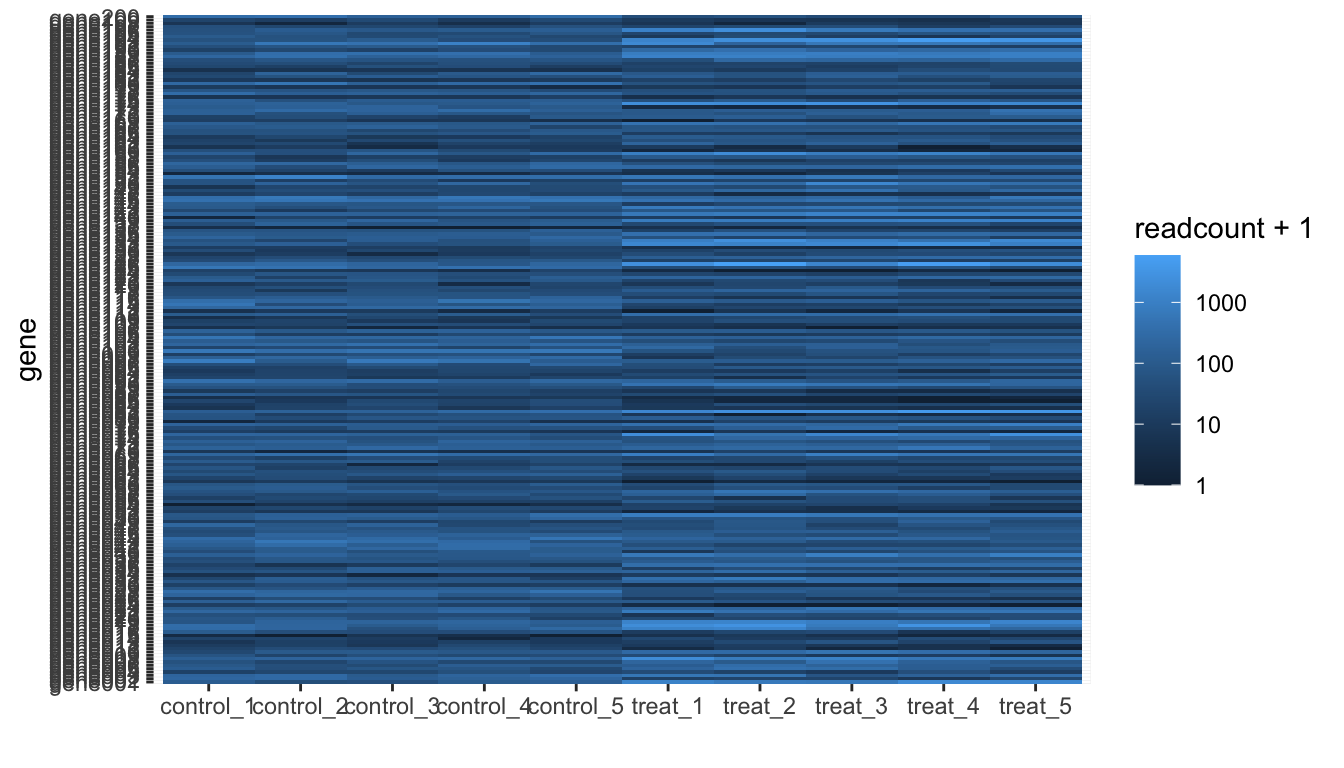
## ヒートマップの色を変更
(gp <- gp + scale_fill_gradient(trans = "log10", low = "grey90", high = "red"))Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale.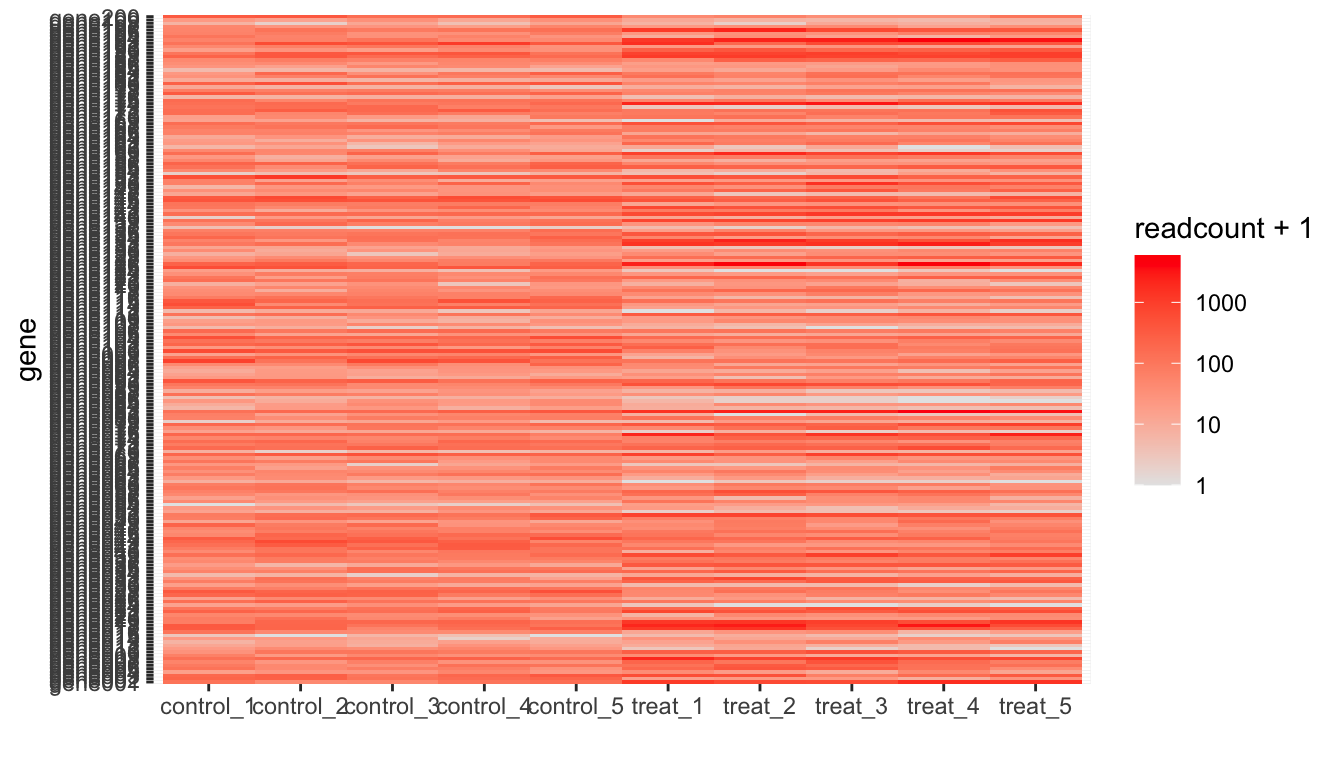
## ヒートマップの余白&縦軸のラベル削除
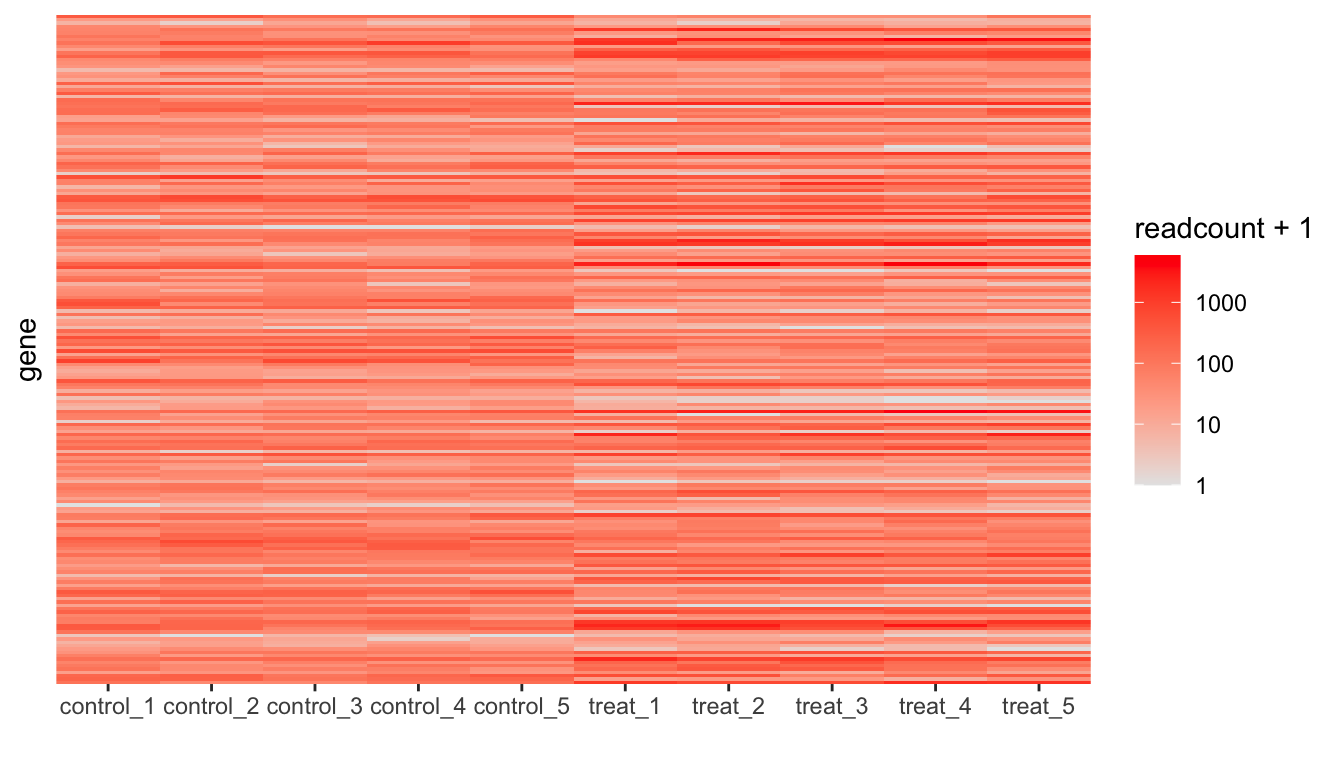
gp +
scale_x_discrete(expand = expansion(0)) +
scale_y_discrete(expand = expansion(0)) +
theme(
axis.ticks.y = element_blank(),
axis.text.y = element_blank()
)
ggplot2でヒートマップを作成すれば、その体裁の整え方はggplot2のやり方になる。 先ほどと同様のデータに少し手を加えて、さらに体裁を整えてみる。
先ほどまでは色分けにリードカウントデータを使っていたが、 これだと発現が低い遺伝子ではパターンの変化が見分けにくい。 そこで、遺伝子のサンプル間における発現量の変化に注目するために、 リードカウントをRPM補正して対数変換し、 遺伝子ごとに正規化(Z変換)してヒートマップを描く。 また、サンプルの条件(対照区か処理区か)でヒートマップを区切ってみやすくする。
このような変更をggplot2だけで行うのは大変なので、 作図用のデータに手を加える(新たに列を加える)ことで対応する。
# データの修正
tbl_plot <-
tbl_exp %>%
# readcountをRPM補正する
purrr::modify_if(is.integer, ~ 1e6 * .x / sum(.x)) %>%
tidyr::pivot_longer(
cols = !c(gene, base, log2fc),
names_to = "sample",
values_to = "RPM"
) %>%
# サンプル条件の列とリピートの列を加える
dplyr::mutate(
condition =
stringr::str_extract(sample, "control|treat") %>%
stringr::str_to_title(),
rep = paste("Rep.", stringr::str_extract(sample, "[12345]"))
) %>%
# 遺伝子ごとに対数変換したRPMのZ-scoreを計算する。
dplyr::mutate(Zscore = scale(log(RPM + 1))[,1], .by = gene)
tbl_plot# A tibble: 2,000 × 8
gene base log2fc sample RPM condition rep Zscore
<chr> <dbl> <dbl> <chr> <dbl> <chr> <chr> <dbl>
1 gene001 87 3.78 control_1 5900. Control Rep. 1 -0.492
2 gene001 87 3.78 control_2 2053. Control Rep. 2 -1.54
3 gene001 87 3.78 control_3 2602. Control Rep. 3 -1.31
4 gene001 87 3.78 control_4 5691. Control Rep. 4 -0.528
5 gene001 87 3.78 control_5 7146. Control Rep. 5 -0.301
6 gene001 87 3.78 treat_1 25424. Treat Rep. 1 0.961
7 gene001 87 3.78 treat_2 13473. Treat Rep. 2 0.330
8 gene001 87 3.78 treat_3 14125. Treat Rep. 3 0.377
9 gene001 87 3.78 treat_4 35484. Treat Rep. 4 1.29
10 gene001 87 3.78 treat_5 32577. Treat Rep. 5 1.21
# ℹ 1,990 more rows# 修正したデータを使ってヒートマップを作図
plot_heatmap <- function(tbl) {
tbl %>%
ggplot(aes(x = rep, y = gene)) +
geom_tile(aes(fill = Zscore)) +
# ここは基本編とおなじ
labs(x = "", y = "") +
scale_x_discrete(expand = expansion(0)) +
scale_y_discrete(expand = expansion(0)) +
# 以下は変更
scale_fill_gradient2() +
facet_grid(cols = vars(condition)) +
theme_linedraw(base_size = 12) +
theme(
axis.ticks = element_blank(),
axis.text.y = element_blank(),
panel.border = element_rect(color = "black", fill = NA),
strip.text = element_text(color = "black", face = "bold"),
strip.background = element_rect(color = "black", fill = NA)
) +
guides(fill = guide_colorbar(
title.theme = element_text(angle = 270),
title = "Z-score transformed RPM",
title.position = "right",
label.position = "left",
barheight = unit(5, "cm"),
barwidth = unit(.3, "cm")
))
}
tbl_plot %>% plot_heatmap()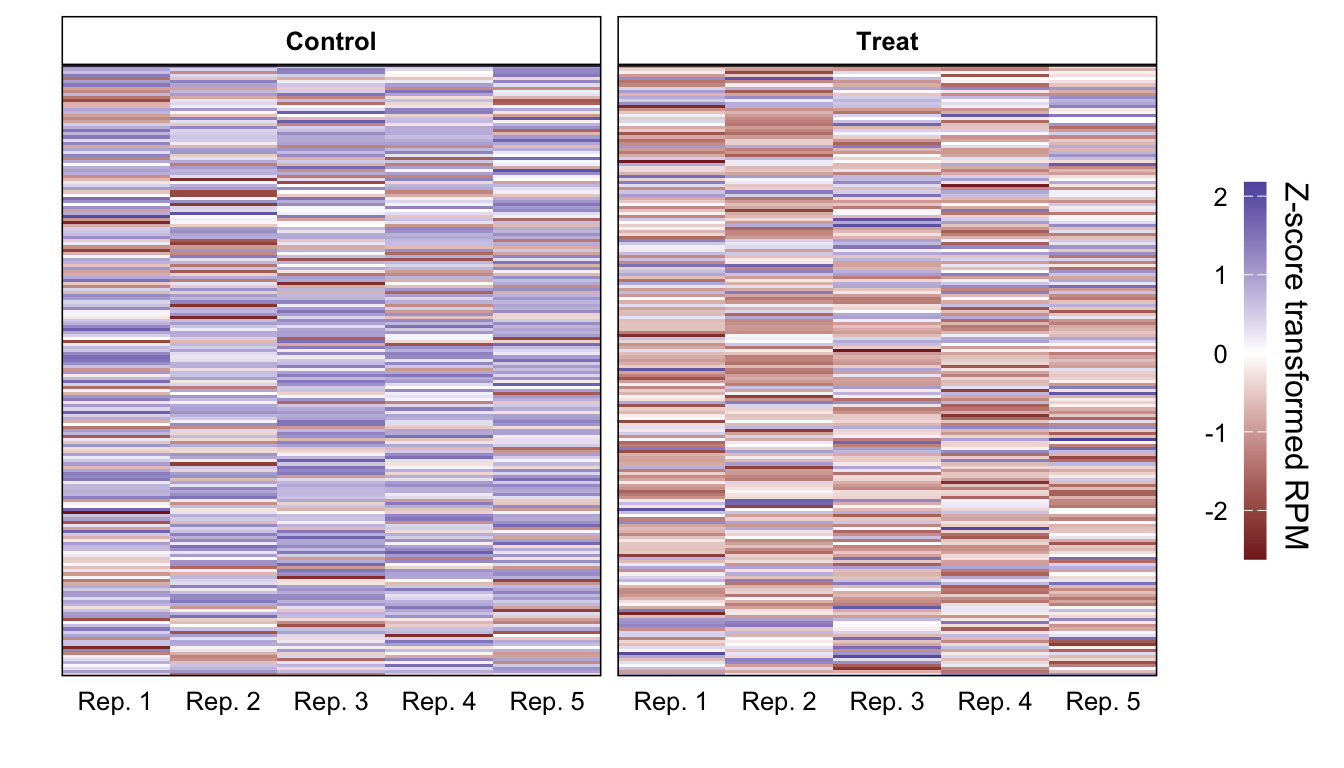
また、遺伝子の順番を何らかの基準で並べ替えることもできる。
ggplot2では離散変数(文字列、character型)の並びは明示的に決めていない場合、 辞書順で因子型(factor型)に変換される。 つまり何も指定しなければ辞書順になる。 そこで、順番を変えたいcharacter型の変数(列)をfactor型に変更し、 明示的に順番を指定すれば良い。 こうした作業にはforcatsパッケージの関数が便利である。
まずは、簡単に発現変動量の値を元に並べ替えをしてみる。
gene_order <- dplyr::arrange(tbl_exp, log2fc)$gene
tbl_plot %>%
dplyr::mutate(gene = forcats::fct_relevel(gene, gene_order)) %>%
plot_heatmap()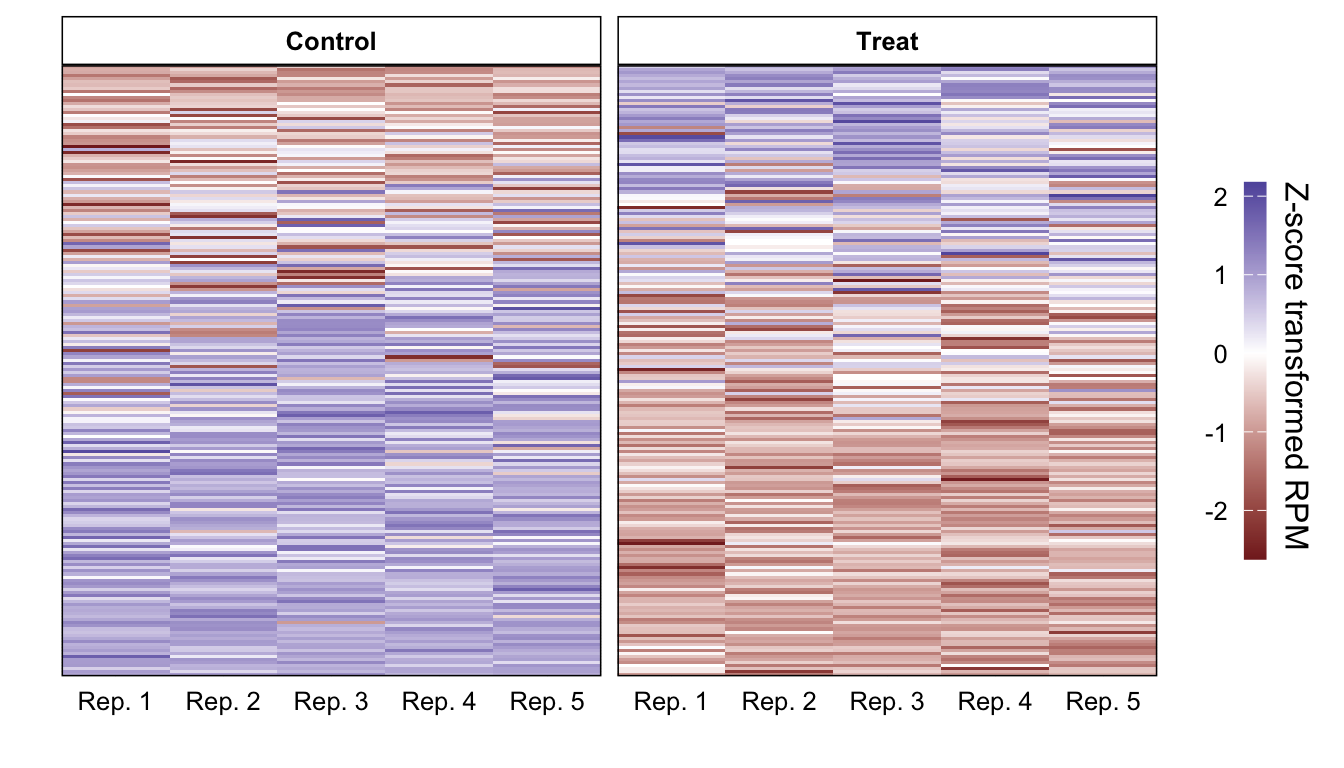
次に、発現パターンを元にクラスタリングを行って、その結果を使って並べ替える。 まずは階層型クラスタリングを行なってみる。 階層型クラスタリングは各要素間の距離をもとに、 アルゴリズムを用いて最も近い要素同士を 一つのクラスタにまとめる作業を繰り返すことで達成される。 Rにはデフォルトで階層型クラスタリングを行うhclust()関数があるのでこれを使うことができる。
hclust()でクラスタリングを行うためには、遺伝子間の発現パターンを元に距離を計算して、距離行列を作成する必要がある。 遺伝子の発現パターンを比較するには相関係数が良いと思うが、 Rの元々ある距離行列を求めるdist()関数ではこれは計算することができない。 そこで、amapパッケージのamap::Dist()関数を用いる。
# 階層型クラスタリング用のdata.frameの準備
df_data <-
tbl_plot %>%
dplyr::select(gene, sample, Zscore) %>%
tidyr::pivot_wider(names_from = sample, values_from = Zscore) %>%
tibble::column_to_rownames("gene")
head(df_data) control_1 control_2 control_3 control_4 control_5 treat_1
gene001 -0.49175909 -1.5416281 -1.3062846 -0.5276727 -0.3012835 0.96135918
gene002 1.01093560 1.0873486 0.7094569 0.8361411 0.9737652 -0.95850613
gene003 0.36384710 0.7501150 -0.3834417 0.3920481 1.1445993 0.65420752
gene004 0.09913786 0.5968042 0.7577385 1.5796809 0.5221213 0.07945511
gene005 0.80995359 -0.6064859 0.2616918 0.1403465 -1.1444680 0.31950414
gene006 0.38003915 -1.0725665 -1.5189545 1.2910079 0.9336676 -0.63042216
treat_2 treat_3 treat_4 treat_5
gene001 0.3295962 0.3766025 1.2930546 1.20801542
gene002 -0.8566026 -0.7540475 -1.4278261 -0.62066516
gene003 0.3846542 -2.1903781 -1.1486152 0.03296381
gene004 -0.9335277 -1.6406241 0.2311051 -1.29189111
gene005 1.2626272 1.1648417 -0.3666657 -1.84134544
gene006 1.3299753 0.3279186 -0.6690319 -0.37163340# 距離として相関係数、アルゴリズムはWard法を使って階層型クラスタリング
hc <-
df_data %>%
amap::Dist(method = "correlation") %>%
hclust(method = "ward.D2")
# クラスタリングした順序を用いてヒートマップを並べ替え
gene_order <- hc$labels[hc$order]
tbl_plot %>%
dplyr::mutate(gene = forcats::fct_relevel(gene, gene_order)) %>%
plot_heatmap()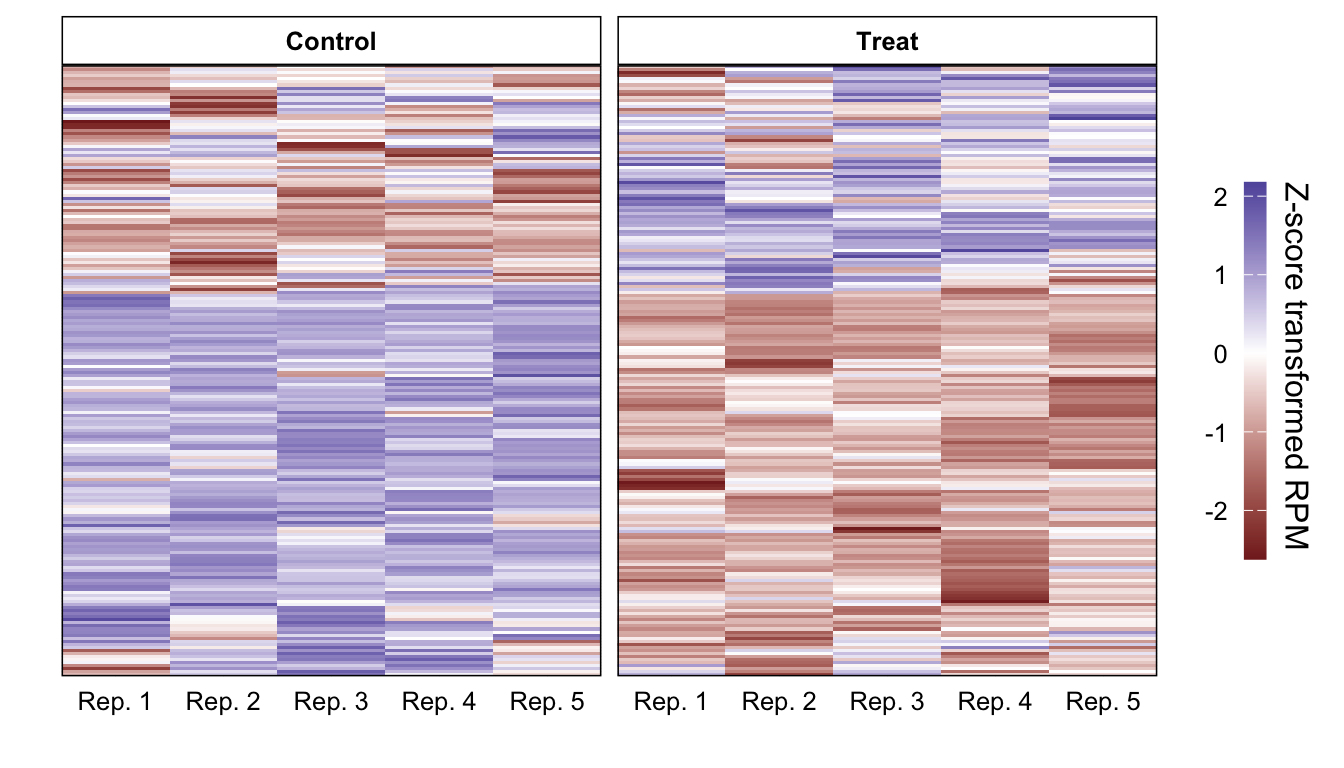
階層型クラスタリングでは、特定のクラスタ数になるように木をある高さで切ることで、 各遺伝子をそれぞれのクラスタに分けることができる。 この操作はcutree()関数で行うことができる。 cutree()でクラスタに分割して、 その情報をもとにクラスタで分けられたヒートマップを作成してみる。
cut_into_cluster <- hc %>% cutree(3)
tbl_plot %>%
dplyr::mutate(cluster = cut_into_cluster[gene]) %>%
dplyr::arrange(cluster) %>%
dplyr::mutate(gene = forcats::fct_inorder(gene)) %>%
plot_heatmap() +
facet_grid(rows = vars(cluster), cols = vars(condition),
scales = "free", space = "free") +
theme(panel.spacing = unit(.5, "mm"))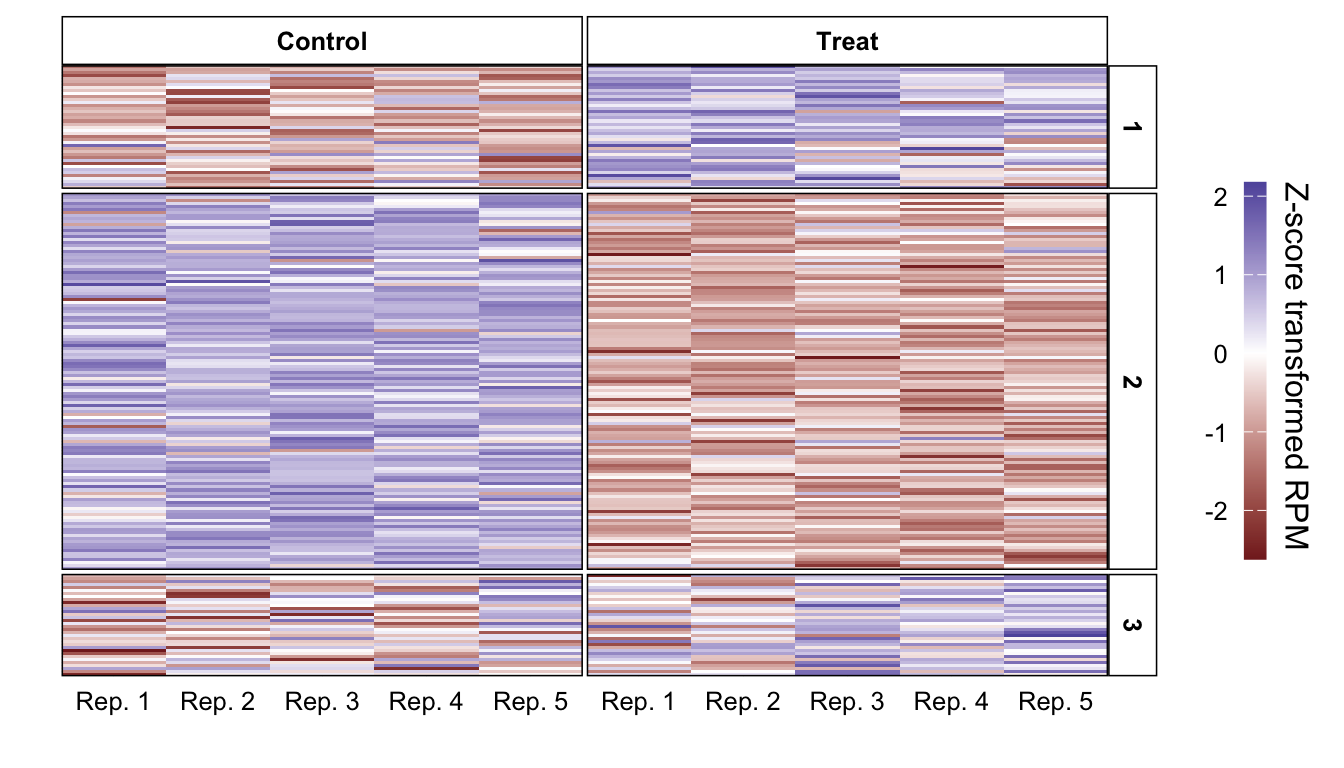
よく使われる非階層型クラスタリングとして、K-mean法によるクラスタリングがある。 Rではkmeans()関数で簡単にクラスタリングできるので、 これを使ってヒートマップをクラスタごとに分割してみる。 また、kmeans()で得られたクラスタの順序はお互いに関連がないので、 各クラスタの中心を元に階層型クラスタリングを行い、 クラスタの並び替えを行う。
set.seed(777)
res_kmean <-
df_data %>%
kmeans(centers = 8)
hc <- hclust(amap::Dist(res_kmean$centers, method = "correlation"))
tbl_plot_w_km_cluster <-
tbl_plot %>%
dplyr::mutate(
cluster =
res_kmean$cluster[gene] %>%
as.character() %>%
forcats::fct_relevel(hc$labels[hc$order])
) %>%
dplyr::arrange(cluster) %>%
dplyr::mutate(gene = forcats::fct_inorder(gene))
tbl_plot_w_km_cluster %>%
plot_heatmap() +
facet_grid(rows = vars(cluster), cols = vars(condition),
scales = "free", space = "free") +
theme(panel.spacing = unit(.5, "mm"))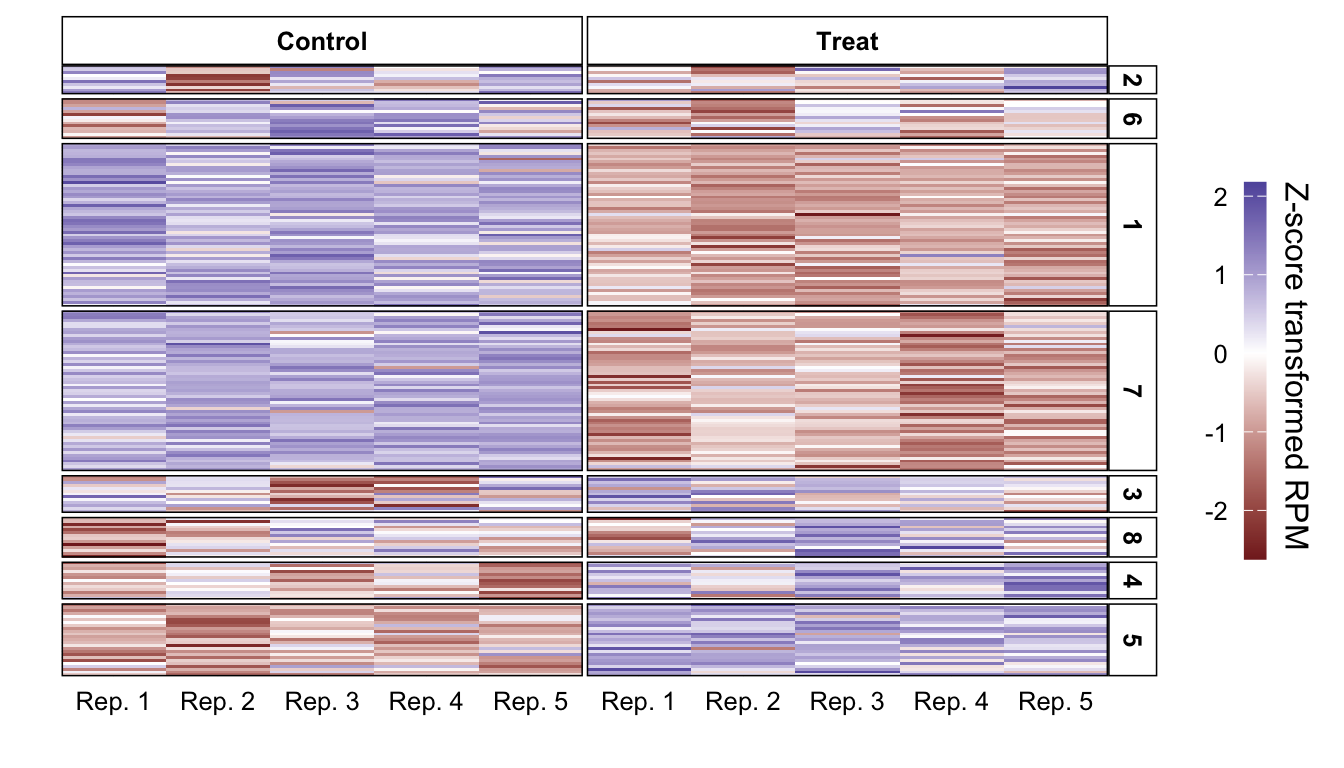
ここまでは階層型クラスタリングに用いたamapパッケージを除いて、 Rをインストールした際に最初からインストールされているパッケージや、 tidyverseのパッケージ群を用いてヒートマップの作図を行なってきた。
これらのパッケージに加えて、 ggplot2の機能をさらに拡張することができるパッケージが多く開発されている。 ここからは、そうした追加のパッケージを用いて、 さらに凝った体裁のヒートマップを作図する方法を紹介する。
ggh4xパッケージを使うと、ネストされたfacetを設定することができる。 これまではリピートをx軸に設定していたが、 “処理条件の中の各反復”という入れ子(ネスト)を、facetとして表現することができる。 今回の例ではそれほどネストさせる意味はないが、 たとえば異なる処理をしたサンプルを経時的に計測したデータなどでは、 ネストされたfacetでヒートマップを作成するとみやすいかもしれない。
gp_hm <-
tbl_plot_w_km_cluster %>%
plot_heatmap() +
facet_grid(rows = vars(cluster), cols = vars(condition),
scales = "free", space = "free", switch = "y") +
theme(
strip.text = element_text(color = "black", face = "bold"),
strip.text.y.left = element_text(color = "black", face = "bold", angle = 90),
strip.background = element_rect(color = NA, fill = NA),
panel.border = element_rect(color = NA, fill = NA),
panel.spacing.x = unit(1.5, "mm"),
panel.spacing.y = unit(1.5, "mm")
)
gp_hm_nested <-
gp_hm +
ggh4x::facet_nested(cols = vars(condition, rep), rows = vars(cluster),
scales = "free", space = "free", nest_line = element_line(), switch = "y") +
theme(
axis.text = element_blank(),
ggh4x.facet.nestline = element_line(colour = "black", linewidth = 1)
)
patchwork::wrap_plots(gp_hm, gp_hm_nested, nrow = 1)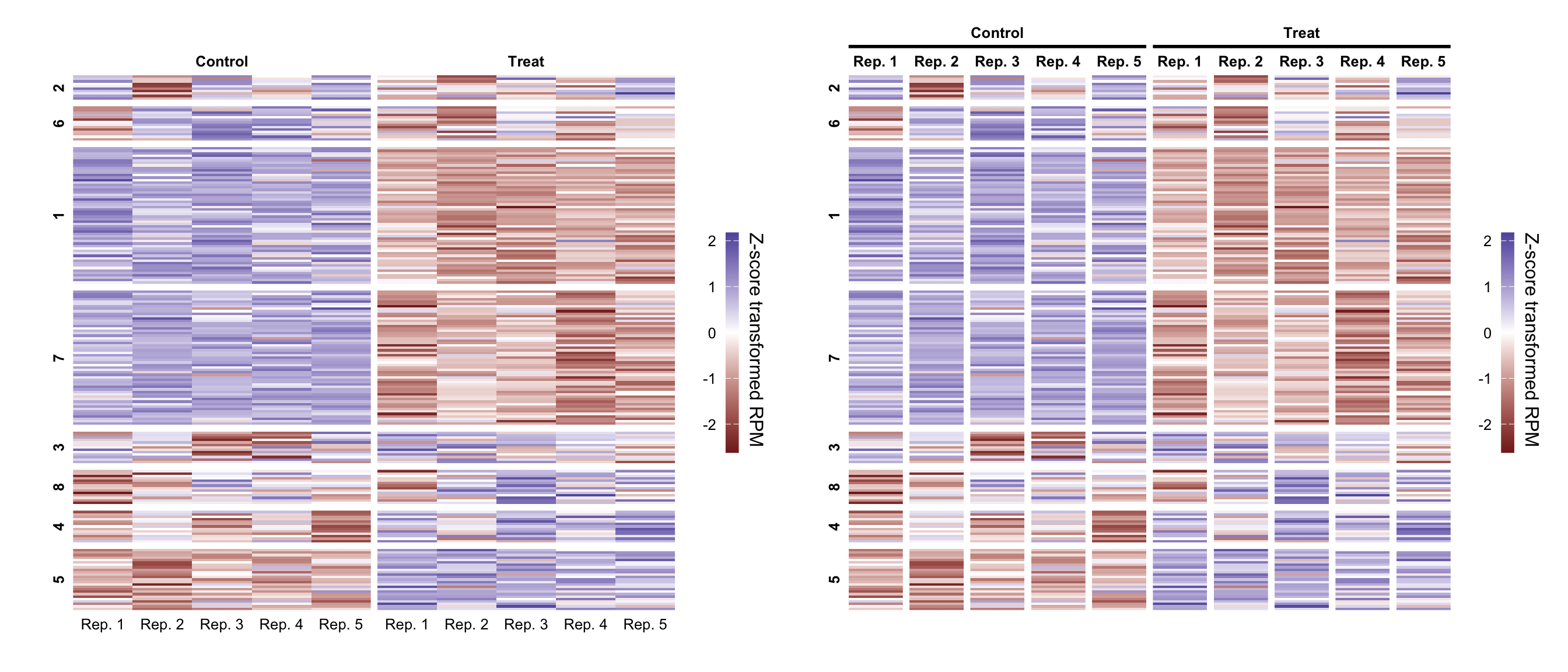
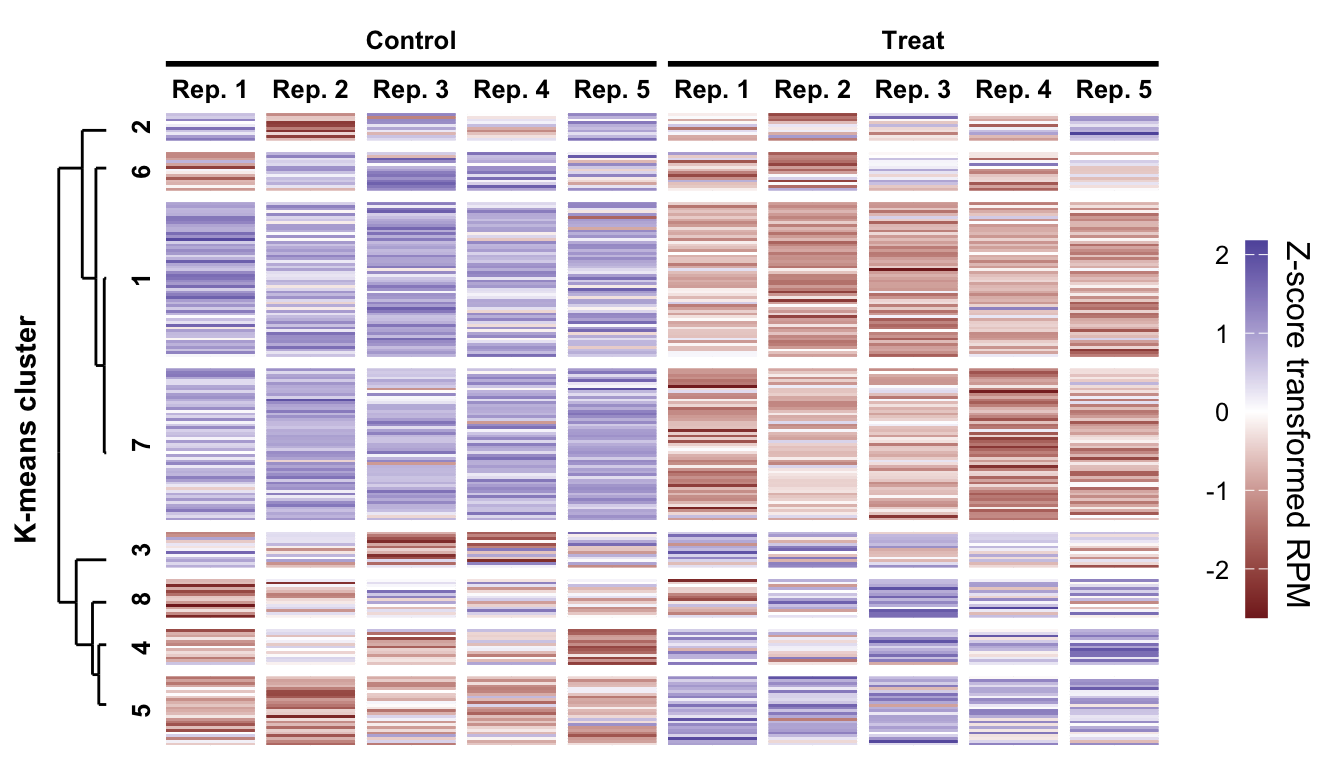
ggdendroパッケージを用いると、階層型クラスタリングを行なったオブジェクトから、 ggplot2で樹形図を描くためのデータを抽出することができる。 これはつまり、ggplot2で樹形図の体裁を整えることができるということである。
また、ggplot2で作成した複数のプロットを一つにまとめるパッケージはいくつかあるので、 そういったものを使えば、ヒートマップの並べ替えの根拠として階層型クラスタリングの 樹形図を組み合わせることができる。 ここでは、ggdendroパッケージとpatchworkパッケージを用いて、 ヒートマップとk-meanで求めた各クラスタの樹形図を組み合わせる方法を紹介する。
ちなみに、共通する軸を持つ複数のプロットを組み合わせる場合は、 各プロットで対応する軸の位置合わせをする必要がある。 うまく位置が合わない場合は、プロットの描画範囲や余白を調整するとよい。
cluster_x_pos <-
res_kmean$cluster %>%
table() %>%
{.[hc$labels[hc$order]]} %>%
{cumsum(.) - (. / 2)}
dend_data <- ggdendro::dendro_data(hc)
# 葉に繋がる横線
tbl_leaf <-
ggdendro::segment(dend_data) %>%
dplyr::filter(x == xend, yend == 0) %>%
dplyr::select(x, xend) %>%
dplyr::mutate(new_x = as.double(cluster_x_pos))
# ノード間の横線
tbl_temp <-
ggdendro::segment(dend_data) %>%
dplyr::filter(x == xend, yend != 0) %>%
dplyr::arrange(y) %>%
dplyr::select(x, xend)
tbl_temp <-
ggdendro::segment(dend_data) %>%
dplyr::filter(y == yend) %>%
dplyr::arrange(desc(y)) %>%
dplyr::slice_head(n = 2) %>%
dplyr::select(x, xend) %>%
unlist() %>%
unique() %>%
sort() %>%
{.[2]} %>%
{tibble::tibble(x = ., xend = .)} %>%
{dplyr::bind_rows(tbl_temp, .)}
for(i in seq_len(nrow(tbl_temp))) {
temp <-
abs(seq_len(nrow(tbl_leaf)) - tbl_temp[i, 1]) %>%
{which(. == min(.))} %>%
{mean(tbl_leaf$new_x[.])} %>%
{dplyr::mutate(tbl_temp[i, ], new_x = .)}
tbl_leaf <- dplyr::bind_rows(tbl_leaf, temp)
}gp_tree <-
ggplot() +
geom_segment(
data =
ggdendro::segment(dend_data) %>%
dplyr::left_join(dplyr::select(tbl_leaf, !xend), by = "x") %>%
dplyr::rename(nx = new_x) %>%
dplyr::left_join(dplyr::select(tbl_leaf, !x), by = "xend") %>%
dplyr::rename(nxend = new_x),
aes(x = y, y = nx, xend = yend, yend = nxend)) +
scale_x_reverse() +
scale_y_reverse(limits = c(200, 0), expand = expansion(add = .5)) +
theme_minimal()
patchwork::wrap_plots(
gp_tree +
labs(y = "K-means cluster") +
theme(
axis.text = element_blank(),
panel.grid = element_blank(),
axis.title.x= element_blank(),
axis.title.y.left = element_text(face = "bold"),
plot.margin = margin()
),
gp_hm_nested +
theme(
axis.title = element_blank(),
plot.margin = margin()
),
nrow = 1, widths = c(.05, .95)
)
sessionInfo()R version 4.3.2 (2023-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Ventura 13.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Asia/Tokyo
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggplot2_3.5.1 magrittr_2.0.3
loaded via a namespace (and not attached):
[1] gtable_0.3.5 jsonlite_1.8.8 dplyr_1.1.4 compiler_4.3.2
[5] tidyselect_1.2.1 stringr_1.5.1 tidyr_1.3.1 scales_1.3.0
[9] yaml_2.3.9 fastmap_1.1.1 amap_0.8-19 ggh4x_0.2.8
[13] R6_2.5.1 patchwork_1.2.0 labeling_0.4.3 generics_0.1.3
[17] knitr_1.48 MASS_7.3-60 htmlwidgets_1.6.4 forcats_1.0.0
[21] tibble_3.2.1 munsell_0.5.1 pillar_1.9.0 rlang_1.1.4
[25] utf8_1.2.4 stringi_1.8.4 xfun_0.46 cli_3.6.3
[29] withr_3.0.0 digest_0.6.34 grid_4.3.2 lifecycle_1.0.4
[33] ggdendro_0.2.0 vctrs_0.6.5 evaluate_0.24.0 glue_1.7.0
[37] farver_2.1.2 fansi_1.0.6 colorspace_2.1-1 rmarkdown_2.25
[41] purrr_1.0.2 tools_4.3.2 pkgconfig_2.0.3 htmltools_0.5.7