igv --version 2>/dev/nullUsing system JDK. IGV requires Java 17.
2.19.1Integrateive Genomics Viewer (IGV) とはゲノムブラウザの一つである。 ゲノムブラウザとは、ゲノム上の遺伝子の位置、遺伝子の構造、ChIP-seqやRNA-seqなどのゲノム上にマッピングできるオミクスデータなどを、 グラフィカルに表示するソフトウェアのことで、 IGVなどを用いることでRNA-seqデータを視覚的に確認することができる。

IGVはGUIで操作することもできるが、簡単なコードを書いて自動で操作することもできる。 今回はIGVのバッチコマンドを利用したコードを書いて、RNA-seqデータのカバレッジデータを表示する方法を解説する。
IGVのアプリケーションは公式ウェブサイトからダウンロードして、インストールすることができる。 Macの場合、コマンドラインからIGVを起動したり、バッチファイルを指定したい場合は、 igvコマンドをインストールする必要があるので、まずはbrewをインストールして、brew install igvでコマンドライン版のIGVをインストールしておく。
コマンドライン版のIGVが正しくインストールできていれば、以下のようにバージョンとコマンドの使用方法が確認できるようになっているはず。
igv --version 2>/dev/nullUsing system JDK. IGV requires Java 17.
2.19.1igv --help 2>/dev/nullUsing system JDK. IGV requires Java 17.
Command line options:
Space delimited list of data files to load
--preferences, -o Path or URL to a preference property file
--batch. -b Path or url to a batch command file
--port, -p IGV command port number (defaults to 60151)
--genome, -g Genome ID (e.g hg19) or path or url to .genome or indexed fasta file
--dataServerURL, -d Path or url to a data server registry file
--genomeServerURL, -u Path or url to a genome server registry file
--indexFile, -i Index file or comma delimited list of index files corresponding to data files
--coverageFile, -c Coverage file or comma delimited list of coverage files corresponding to data files
--name, -n Name or comma-delimited list of names for tracks corresponding to data files
--locus, -l Initial locus
--header, -H http header to include with all requests for list of data files
--igvDirectory Path to the local igv directory. Defaults to <user home>/igv
--version Print the IGV version and exit
--help Print this output and exitIGVアプリを起動すると、左上のメニューから自分が使いたい生物のゲノムデータを選ぶことができる。
今回は シロイヌナズナ (A. thaliana) を使いたいのだが、IGVの公式が提供しているゲノム情報と、 個人的に解析で使用しているゲノム情報とで配列名などが異なり以降の作業で困るので、 自分のPC上に保存しているゲノム配列のファイルを指定する方法を紹介する。
まずは自分のPC上に使用したいゲノム配列のデータを保存する。 ここではpath/to/fasta/というフォルダにシロイヌナズナのゲノム配列のFASTAファイルを保存したとする。 FASTAファイルをIGVで使用するにはインデックスが必要なので、ない場合は作成する。
あとは、準備したFASTAファイルをコマンドラインから以下のように指定することで、ゲノムを指定してIGVを起動することができる。
igv --genome path/to/fasta/Athaliana_allN.fa遺伝子構造の情報は公開されているGTFファイルなどを、IGVで読み込むことでみることができる。 ここではpath/to/gtf/というフォルダに、必要なGTFファイルを保存した。
grep -e "AT1G010[123456]0" path/to/gtf/TAIR10_GFF3_genes_transposons.gtf > path/to/gtf/TAIR10_GFF3_genes_transposons_mini.gtfまた、RNA-seqデータのカバレッジデータを見たい場合はBAMファイルを読み込むか、 より軽量なbigwig形式のデータを読み込むことで、見ることができるようになる。 ここでは、path/to/coverage/というフォルダにbigwigファイルを保存した。
IGVは以下のページにリストアップされたバッチコマンドが書かれた バッチスクリプトファイルを使って動作を制御することができる。
Integrative Genomics Viewer. Fast, efficient, scalable visualization tool for genomics data and annotations - igvteam/igv
 https://github.com/igvteam/igv/wiki/Batch-commands
https://github.com/igvteam/igv/wiki/Batch-commands
今回はIGVのバッチコマンドを使って以下の作業を自動化するスクリプトを作成してみる。
bigwigファイル)をtrackに追加するこの作業を自動で実行するバッチファイルを作成したものがこちら。
path/to/example.batch
new
snapshotDirectory path/to/snapshots
goto Chr1:100-200
load path/to/gtf/TAIR10_GFF3_genes_transposons_mini.gtf
setTrackHeight 300 TAIR10_GFF3_genes_transposons_mini.gtf
expand TAIR10_GFF3_genes_transposons_mini.gtf
load path/to/coverage/sampleA_1_mini.bw
setColor #363636 sampleA_1_mini.bw
setDataRange auto sampleA_1_mini.bw
setTrackHeight 100 sampleA_1_mini.bw
load path/to/coverage/sampleB_1_mini.bw
setColor #F63636 sampleB_1_mini.bw
setDataRange auto sampleB_1_mini.bw
setTrackHeight 100 sampleB_1_mini.bw
goto AT1G01010
snapshot AT1G01010.png
exitこのバッチファイルでやっていることを解説すると、
expandするときに領域が広すぎると問題になるので事前に狭い位置を指定しておくbigwiggle形式のRNA-seqのカバレッジデータを読み込んでカバレッジのtrackを作成auto (描画範囲から自動で設定)となる。 ゲノムデータとこのバッチファイルを指定して、 コマンドラインからIGVを実行するには以下のようにすればよい。
igv --genome path/to/fasta/Athaliana_allN.fa --batch path/to/example.batch実行が終わると、path/to/snapshots/ディレクトリに以下のようにスナップショットが生成される。

数個の遺伝子、あるいは領域についてスナップショットを作成するバッチファイルを作成してもあまり便利ではないが、 バッチファイルを作成する利点はたくさんスナップショットを撮りたいときにある。
次はカバレッジデータが複数あって、かつ複数の遺伝子についてスナップショットを撮りたい場合を考えてみよう。 先ほどのバッチスクリプトをベースにして、カバレッジデータのtrackを指定する部分と、 遺伝子領域に移動してスナップショットを撮る部分を、 ファイル毎、遺伝子毎に書き換えればうまくいくだろう。
このようなスクリプトを部分的に変更して、繰り返し処理を行うコードを生成するには、 Rのglueパッケージが非常に有用である。
Rのglueパッケージを使って、他の言語の一部分だけ変更したスクリプトを生成する。
https://t-arae.blog/posts/2024/2024-08-18-glue-shell-script/

上の記事で説明したことを応用して、Rを使ってバッチスクリプトを生成してみよう。
### 1. テンプレートを作成する ###
tmpl_header <- r"(new
snapshotDirectory {dir_output}
goto Chr1:100-200
load {gtf_file}
setTrackHeight 300 {gtf_file}
expand {gtf_file}
)"
# bigwiggleファイルを読み込んでカバレッジを設定する部分
tmpl_load_coverage <- r"(load {bw_file}
setColor {color} {track}
setDataRange auto {track}
setTrackHeight 100 {track}
)"
# 遺伝子領域に移動してスナップショットを撮影する部分
tmpl_snapshot <- r"(goto {region}
snapshot {gene_id}.png
)"
tmpl_footer <- r"(exit
)"
### 2. 置換するテキストデータを準備する。 ###
dir_output <- "path/to/snapshots2"
gtf_file <- "path/to/gtf/TAIR10_GFF3_genes_transposons_mini.gtf"
# bigwiggleファイルのデータ
tbl_tracks <- tibble::tribble(
~bw_files, ~colors,
"path/to/coverage/sampleA_1_mini.bw", "#363636",
"path/to/coverage/sampleA_2_mini.bw", "#363636",
"path/to/coverage/sampleB_1_mini.bw", "#F63636",
"path/to/coverage/sampleB_2_mini.bw", "#F63636",
)
# 遺伝子領域のデータ
tbl_genes <- tibble::tribble(
~gene_ids, ~regions,
"AT1G01010", "Chr1 3631 5899",
"AT1G01020", "Chr1 6788 9130",
"AT1G01030", "Chr1 11649 13714",
"AT1G01040", "Chr1 23416 31120",
"AT1G01050", "Chr1 31170 33171",
"AT1G01060", "Chr1 33662 37840",
)
### 3. glueパッケージを使ってテキストを置換し、バッチスクリプトのパーツを作成する ###
temp_cmds <- glue::glue(tmpl_header, .trim = FALSE)
# カバレッジファイル読み込みのスクリプト
for(i in seq_len(nrow(tbl_tracks))) {
bw_file <- tbl_tracks$bw_files[i]
color <- tbl_tracks$colors[i]
track <- fs::path_file(bw_file)
temp_cmds <- c(temp_cmds, glue::glue(tmpl_load_coverage, .trim = FALSE))
}
# スナップショット撮影のスクリプト
for(i in seq_len(nrow(tbl_genes))) {
gene_id <- tbl_genes$gene_ids[i]
region <- tbl_genes$regions[i]
temp_cmds <- c(temp_cmds, glue::glue(tmpl_snapshot, .trim = FALSE))
}
temp_cmds <- c(temp_cmds, glue::glue(tmpl_footer, .trim = FALSE))
### 4. 作成したバッチスクリプトのパーツをファイルに出力 ###
temp_cmds |> readr::write_lines("path/to/example2.batch")このRのコードで出力されるIGVのバッチスクリプトは以下のようになる。
path/to/example2.batch
new
snapshotDirectory path/to/snapshots2
goto Chr1:100-200
load path/to/gtf/TAIR10_GFF3_genes_transposons_mini.gtf
setTrackHeight 300 path/to/gtf/TAIR10_GFF3_genes_transposons_mini.gtf
expand path/to/gtf/TAIR10_GFF3_genes_transposons_mini.gtf
load path/to/coverage/sampleA_1_mini.bw
setColor #363636 sampleA_1_mini.bw
setDataRange auto sampleA_1_mini.bw
setTrackHeight 100 sampleA_1_mini.bw
load path/to/coverage/sampleA_2_mini.bw
setColor #363636 sampleA_2_mini.bw
setDataRange auto sampleA_2_mini.bw
setTrackHeight 100 sampleA_2_mini.bw
load path/to/coverage/sampleB_1_mini.bw
setColor #F63636 sampleB_1_mini.bw
setDataRange auto sampleB_1_mini.bw
setTrackHeight 100 sampleB_1_mini.bw
load path/to/coverage/sampleB_2_mini.bw
setColor #F63636 sampleB_2_mini.bw
setDataRange auto sampleB_2_mini.bw
setTrackHeight 100 sampleB_2_mini.bw
goto Chr1 3631 5899
snapshot AT1G01010.png
goto Chr1 6788 9130
snapshot AT1G01020.png
goto Chr1 11649 13714
snapshot AT1G01030.png
goto Chr1 23416 31120
snapshot AT1G01040.png
goto Chr1 31170 33171
snapshot AT1G01050.png
goto Chr1 33662 37840
snapshot AT1G01060.png
exit実行してみると、path/to/example2.batchにスナップショットが複数生成される。
igv --genome path/to/fasta/Athaliana_allN.fa --batch path/to/example2.batch





今回はスナップショットを撮る遺伝子、領域のデータを自分で用意したが、 実際にやる場合は何らかの解析結果のCSVファイルなどを読み込んで、 遺伝子のリストを作成し、それに対してバッチスクリプトを生成することになるだろう。
最後に選択的スプライシングで特定の位置のカバレッジがサンプル間で異なる場合に、 特定の領域を強調表示してスクリーンショットを撮ってみよう。 これには、regionコマンドを使うことができる。
path/to/example3.batch
new
snapshotDirectory path/to/snapshots3
goto Chr1:100-200
load path/to/gtf/TAIR10_GFF3_genes_transposons_mini.gtf
setTrackHeight 300 TAIR10_GFF3_genes_transposons_mini.gtf
expand TAIR10_GFF3_genes_transposons_mini.gtf
load path/to/coverage/sampleA_1_mini.bw
setColor #363636 sampleA_1_mini.bw
setDataRange auto sampleA_1_mini.bw
setTrackHeight 100 sampleA_1_mini.bw
load path/to/coverage/sampleB_1_mini.bw
setColor #F63636 sampleB_1_mini.bw
setDataRange auto sampleB_1_mini.bw
setTrackHeight 100 sampleB_1_mini.bw
goto Chr1:6788-9130
region Chr1 8571 8594
snapshot AT1G01020.png
exitigv --genome path/to/fasta/Athaliana_allN.fa --batch path/to/example3.batch
第一エキソン末端にあるドナーサイトが異なる領域を強調してみた。 カバレッジの上に赤い四角が表示され、縦線で強調したい領域を目立たせることができている。
今回の可視化に作業に使用したファイルをまとめたファイル(archive.tar.gz)
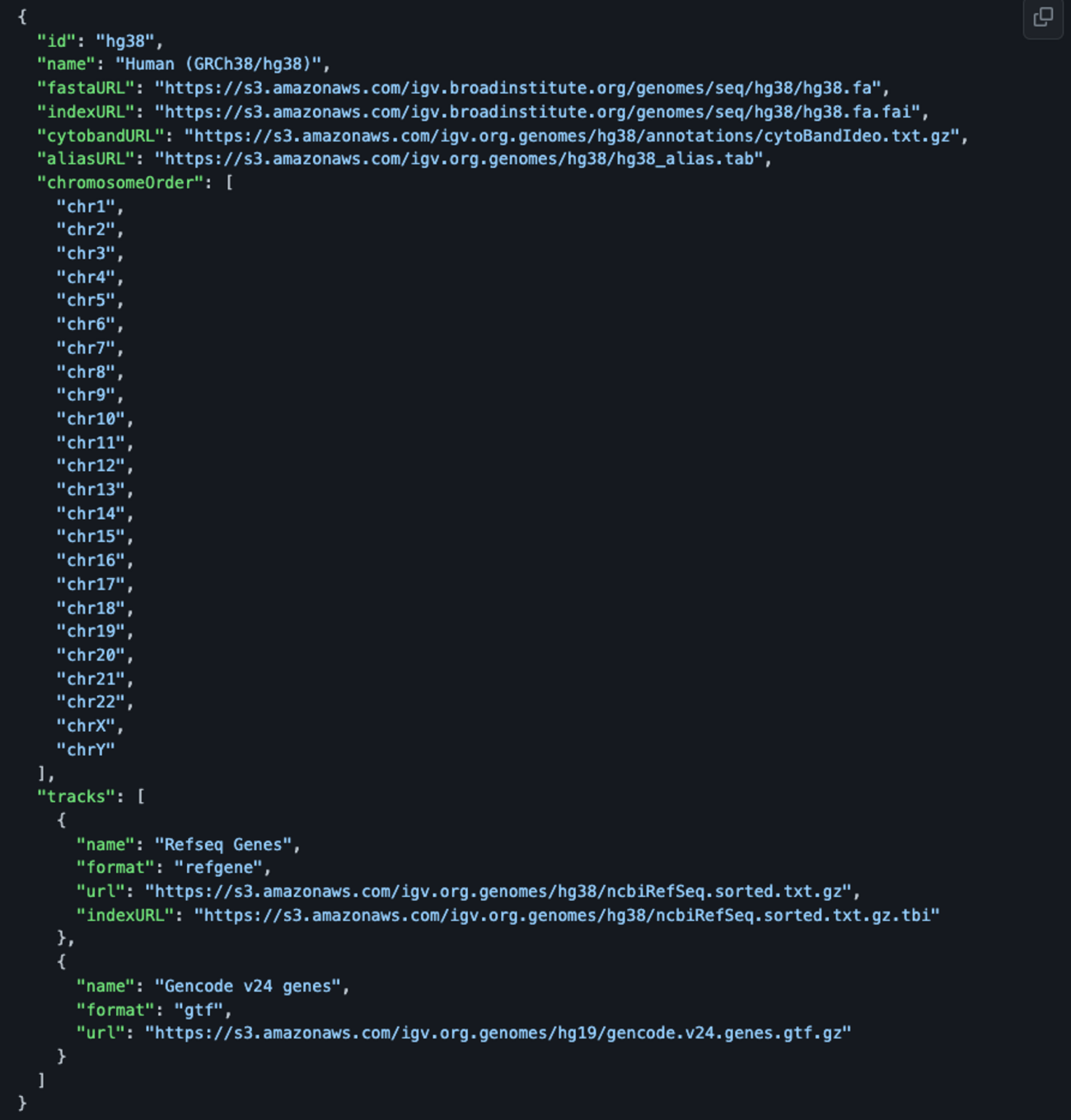
以前書いたIGVのtipsという記事に時々アクセスがあるようなので、今日は再びゲノムブラウザIGVのtipsの紹介をします。 (自分のNGS講習資料(2023・フィリピン大のラボ)より) 1、JSONファイルでの読み書き IGVはゲノムファイルやアノテーションをGenomeメニューから読み込むことよりもJSON形式でゲノムを指定する事を推奨しています。以下の形式になります。 https://github.com/igvteam/igv/wiki/JSON-Genome-Format (manualより転載) アノテーション、cytobandなどのファイルはホストされているURLまたはローカルマ…
https://kazumaxneo.hatenablog.com/entry/2023/03/31/022058
sessionInfo()R version 4.4.1 (2024-06-14)
Platform: aarch64-apple-darwin20
Running under: macOS Ventura 13.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Asia/Tokyo
tzcode source: internal
attached base packages:
[1] stats graphics grDevices datasets utils methods base
loaded via a namespace (and not attached):
[1] crayon_1.5.3 vctrs_0.6.5 cli_3.6.3 knitr_1.49
[5] rlang_1.1.4 xfun_0.50 renv_1.0.11 jsonlite_1.8.9
[9] glue_1.8.0 bit_4.5.0.1 htmltools_0.5.8.1 hms_1.1.3
[13] rmarkdown_2.29 evaluate_1.0.3 tibble_3.2.1 tzdb_0.4.0
[17] fastmap_1.2.0 yaml_2.3.10 lifecycle_1.0.4 compiler_4.4.1
[21] fs_1.6.5 pkgconfig_2.0.3 digest_0.6.37 R6_2.5.1
[25] readr_2.1.5 tidyselect_1.2.1 parallel_4.4.1 vroom_1.6.5
[29] pillar_1.10.1 magrittr_2.0.3 tools_4.4.1 bit64_4.5.2